中国北方常见芫菁分子物种界定(鞘翅目:,芫菁科)
程海云, 段家充, 张 超, 潘 昭
(河北大学生命科学学院, 生命科学与绿色发展研究院, 河北省动物系统学与应用重点实验室, 河北保定 071002)
芫菁科(Meloidae)隶属于鞘翅目(Coleoptera)拟步甲总科(Tenebrionoidea),世界已知3亚科16族133属近3 000种,广泛分布于除新西兰、南极和波利尼西亚群岛之外的世界各地;
中国记录2亚科8族27属近200种(亚种),各省市自治区均有记载(Bolognaetal., 2010; Pan and Ren, 2020)。该科昆虫是一类重要的资源昆虫,因其复变态等独特的生物学特性而被学者广泛关注,尤其是斑蝥素的药用价值更是近年芫菁应用研究的热点之一(杨玉霞和任国栋, 2005; Bolognaetal., 2010)。然而,芫菁科部分物种的外部形态特征十分近似,在形态分类研究中经常出现难以区分、错误鉴定的情况,如北方常见物种圆点斑芫菁Mylabrisaulica和苹斑芫菁Mylabriscalida(Pan and Ren, 2020)、横纹沟芫菁Hycleussolonicus(Panetal., 2017)等。分子系统学的发展,特别是DNA条形码的提出和兴起,为解决以上问题提供了可行的方案。Liu等(2016)基于线粒体COI基因片段序列,对西北豆芫菁Epicautasibirica、疑豆芫菁Epicautadubia和中华豆芫菁Epicautachinensis分类研究的有效性进行了探讨,提出后两者是西北豆芫菁的次异名,然而该研究并未使用分子物种界定方法,仅是基于分支单系性得出的结论。
为更加全面地评估分子物种界定方法在芫菁科分类研究中的可行性,本研究以中国北方常见芫菁科昆虫为研究材料,选取线粒体COI基因片段和核CAD基因片段序列,分别采用自动条形码间隔探索(automatic barcode gap discovery, ABGD)、广义混合Yule溯祖模型(generalized mixed Yule coalescent,GMYC)、贝叶斯泊松树进程(Bayesian Poisson tree processes, bPTP)和贝叶斯系统发育和系统地理分析(Bayesian phylogenetics and phylogeography, BPP)方法进行分子物种界定。在对比形态鉴定结果的基础上,探讨以上序列和分析方法界定的准确性和适用性,以期为芫菁科的整合分类研究提供数据支持。
1.1 样本选取
本研究以中国北方常见芫菁科昆虫为研究对象,选取58个样本,经形态鉴定,共涉及2亚科4族6属18种;
选择拟步甲科(Tenebrionidae)黄粉虫Tenebriomolitor为外群。实验样本大多来自河北大学博物馆馆藏,少部分样本序列下载自GenBank,详见表1。

1.2 形态鉴定
选取样本均使用Nikon SMZ1500立式显微镜进行形态学鉴定。形态鉴定参考中国芫菁分类相关成果,如:Pan等(2010, 2017),潘昭等(2010a, 2010b, 2011, 2013), Li等(2020)以及Pan和Bologna(2021)。
1.3 DNA的提取、PCR扩增及测序
从酒精泡制标本中取后足肌肉组织,使用昆虫基因组DNA提取试剂盒(倍沃医学科技)提取总DNA。通过PCR扩增COI和CAD基因片段,COI基因扩增引物为TW-N1284(5′-AAACTAACARCCTTC AAAGYTGT-3′)和COI-R2329(5′-ACHGTAAAYATA TGATGGGCTCA-3′),反应程序:94℃预变性3 min;
94℃变性1 min, 48℃延伸1.5 min, 40个循环;
72℃复性1.5 min;
72℃最终延伸3 min。CAD基因扩增引物为CD439F(5′-TTCAGTGTACARTTYCAYC CHGARCAYAC-3′)和CD688R(5′-TGTATACCTAG AGGATCDACRTTYTCCATRTTRCA-3′),反应程序:
94℃预变性3 min;
94℃变性30 s, 55℃延伸30 s, 50个循环;
72℃复性1 min;
72℃最终延伸3 min(Wild and Maddison, 2008)。PCR扩增采用25 μL体系:上下游引物(10 μmol/L)各1 μL,去离子水8.5 μL, PCR Mix 12.5 μL,DNA模板2 μL。
PCR产物经1%琼脂糖凝胶电泳检测,送通用生物系统(安徽)有限公司进行双向测序。
1.4 基因序列处理和系统发育分析
所得序列使用DNASTAR SeqMan v7.1.0进行检查和拼接,在NCBI中进行BLAST分析,确定所得序列是否为目的序列, 使用MEGA X (Kumaretal., 2018)中的Clustal W进行序列的比对。所有新得到的序列上传至GenBank(登录号见表1)。
使用MEGA X分别对COI和CAD序列数据集进行序列碱基含量分析及种内和种间遗传距离分析(Kumaretal., 2018)。使用PhyloSuite v1.2.2 (Zhangetal., 2020)将COI和CAD序列数据集串联,构建3个序列数据集,即COI,CAD和COI+CAD串联序列数据集。使用PhyloSuite内嵌的MrBayes 3.2(Ronquistetal., 2012),使用贝叶斯推断法(Bayesian inference, BI),分别基于3个数据集重建系统发育树,运行2 000 000世代,采样频率为每100代一次,确保分裂频率分支频率的标准偏差小于0.01(Ronquist and Huelsenbeck, 2003),以获得高支持率的合意树,生成合意树之前舍弃25%的老化样本,最佳碱基替代模型使用ModelFinder (Kalyaanamoorthyetal., 2017)进行评估。
最后,使用Evolview v3 (Subramanianetal., 2019)对系统树进行可视化编辑。
1.5 物种界定分析
基于3个数据集,分别使用自动条形码间隔探索(ABGD),广义混合Yule溯祖模型(GMYC),贝叶斯泊松树进程(bPTP)和贝叶斯系统发育和系统地理分析(BPP)方法进行物种界定分析,具体参数设置如下:
ABGD:使用ABGD在线分析(https:∥bioinfo.mnhn.fr/abi/public/abgd/abgdweb.html)(金倩等, 2017),设置参数X(relative gap width)为0.5,其他参数为默认值,将得到的序列划分情况与形态鉴定的结果进行比较分析。
GMYC(金倩等, 2017):基于BEAST v1.10.4 (Drummond and Rambaut, 2007)构建超度量二歧分支树(有根),选择严格分子钟。MCMC 链为10 000万代,每1 000代参数取样一次,Burn-In默认为链长10%,使用Tracer v1.7.2(Rambautetal., 2018)评估系统树的收敛情况。将含有树文件的*.trees.txt 文件导入到Tree Annotator v1.10.4生成所需系统树。Burn In (as trees)根据 Tracer v1.7.2分析结果(Burn-In 链长/采样频率)设置为1 000,最后生成所需最大分支可信度树(maximum clade credibility tree),导入GMYC进行分析。
bPTP(Zhangetal., 2013):本实验使用PTP的更新版本bPTP进行界定分析,将GMYC分析中得到的Newick系统树上传到PTP在线网站(https:∥species.h-its.org/ptp/)进行分析,设置为有根树,输入外群名称,马尔科夫链代数为500 000,其他参数为默认值。
BPP(Yang, 2015):基于串联基因数据集构建的贝叶斯系统树,在BPP v4.2.9中利用A11模板输入指导树(基于COI序列构建的BI树),运行分析。
2.1 基因序列特征
COI基因片段序列经拼接、比对、剪切处理后的长度为561 bp。使用MEGA X软件中的Kimura双参数模型(Kimura 2-parameter model)进行序列碱基含量分析及种内、种间遗传距离分析(表2和3),结果显示:序列的T, C, A和G平均含量分别为33.5%, 22.3%, 26.8%和17.5%,平均A+T含量显著高于G+C含量,变异多发生在碱基第3位;
Hycleussolonicus,Mylabrisaulica,Epicautamegalocephala,Mylabrissibirica,Lyttacaraganae,Meloeproscarabaeus,Meloeviolaceus和Epicautasibirica的种内平均遗传距离分别为0.92%, 0.71%, 0.83%, 0.30%, 1.66%, 1.34%, 0.11%和0.24%,其余10个物种样本量不足,无法进行种内遗传距离分析;
18种芫菁的种间遗传距离在9.49%~22.10%之间,平均值为18.36%。
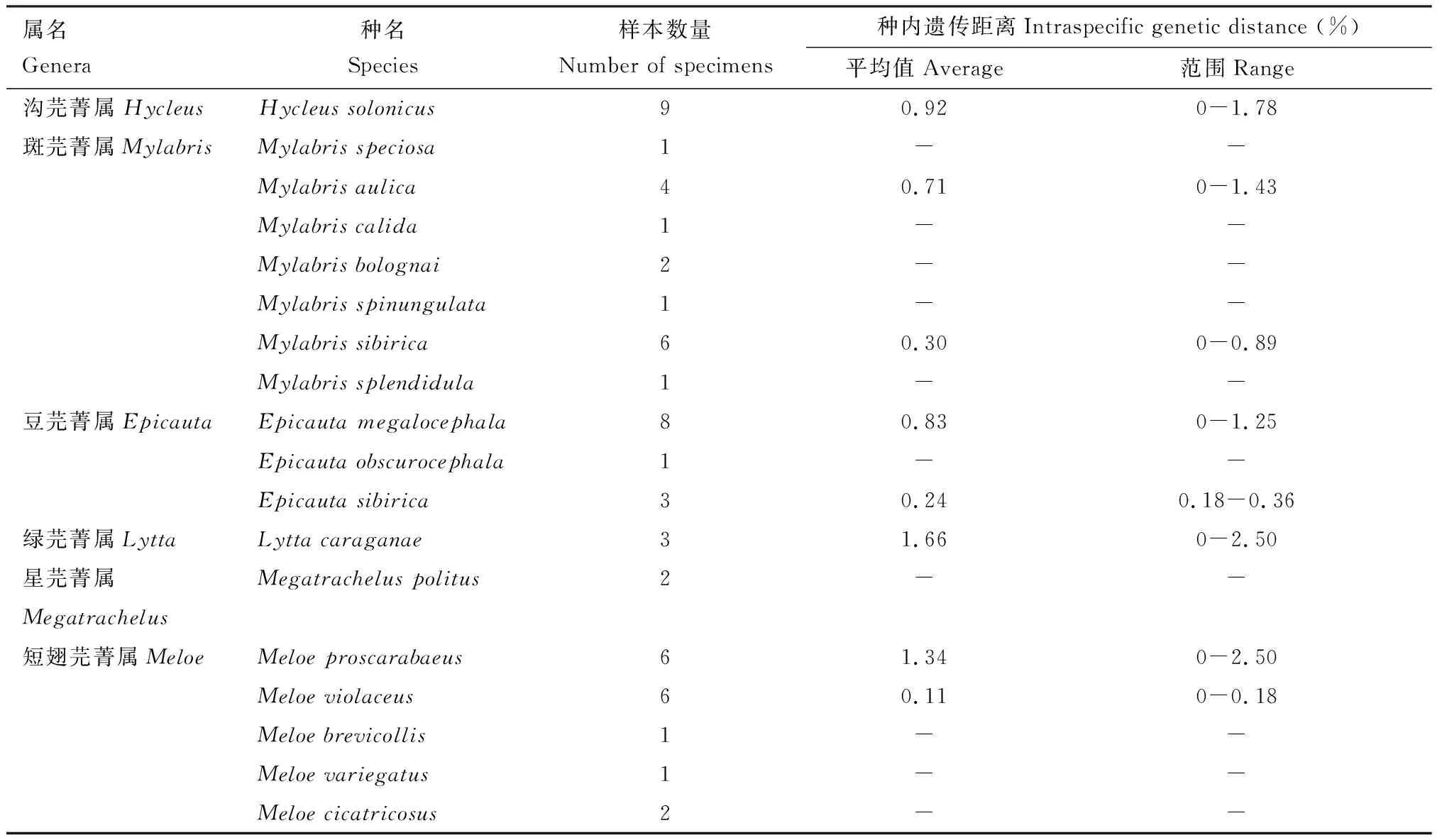
表2 基于COI基因片段序列计算得出芫菁科种内平均遗传距离和种内遗传距离范围Table 2 Average intraspecific genetic distance and range of intraspecific genetic distance of Meloidaecalculated based on COI fragment sequences
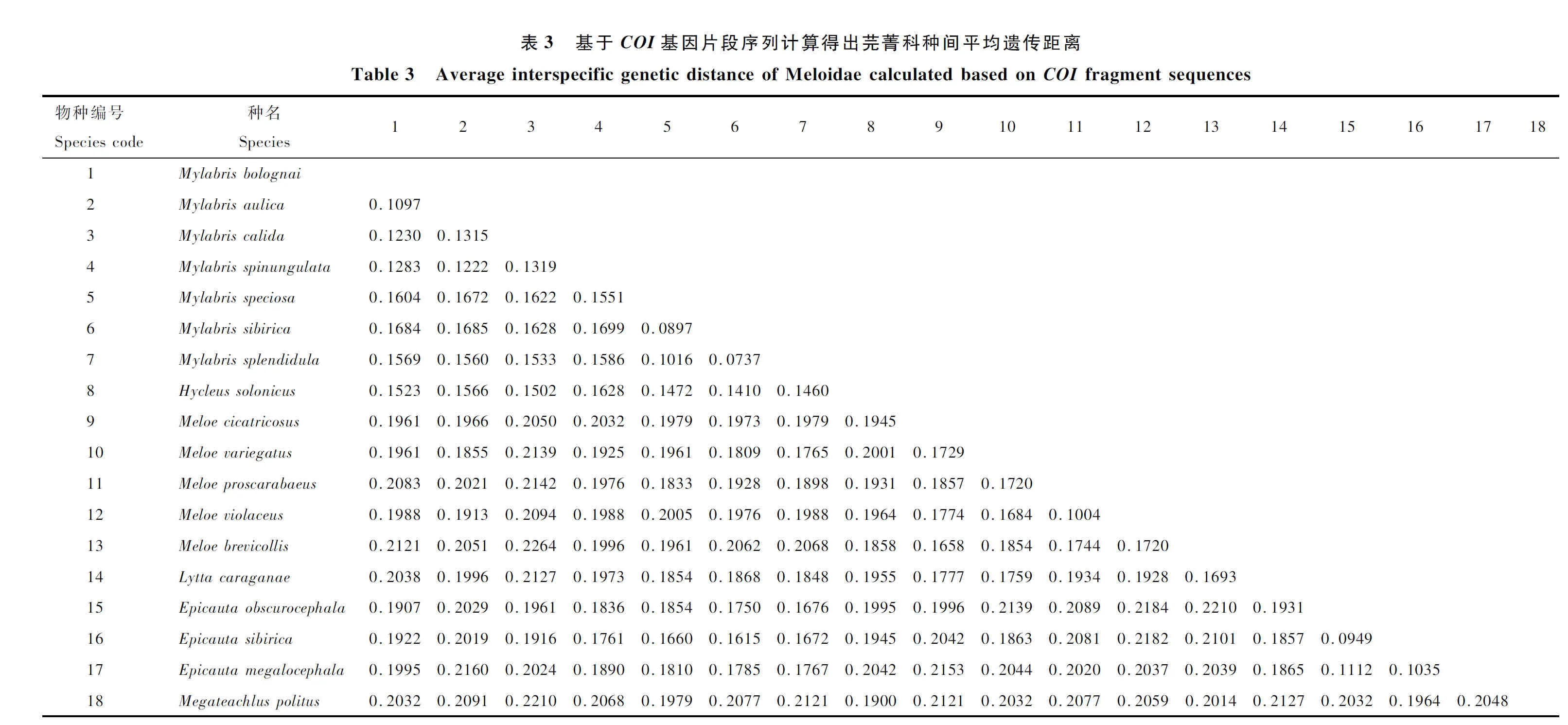
CAD基因片段序列处理后长度为730 bp。使用MEGA X 软件中的Kimura 双参数模型进行序列碱基含量分析及种内种间遗传距离分析(表4和5),结果显示:T, C, A和G平均含量分别为33.5%, 22.3%, 26.8%和17.5%,平均A+T含量显著高于G+C含量,没有碱基变异偏向性,密码子的1, 2和3位均有发生变异;
Hycleussolonicus,Mylabrisaulica,Epicautamegalocephala,Epicautasibirica,Lyttacaraganae,Meloeproscarabaeus和Meloeviolaceus的种内平均遗传距离为1.71%, 0.65%, 0.91%, 0.73%, 0.46%, 0.27%和0.06%,Mylabrissibirica没有变异,其余10个物种样品量不足,无法进行种内遗传距离分析; 18种芫菁的种间遗传距离是处于0.43%~22.33%之间,平均值为13.85%。
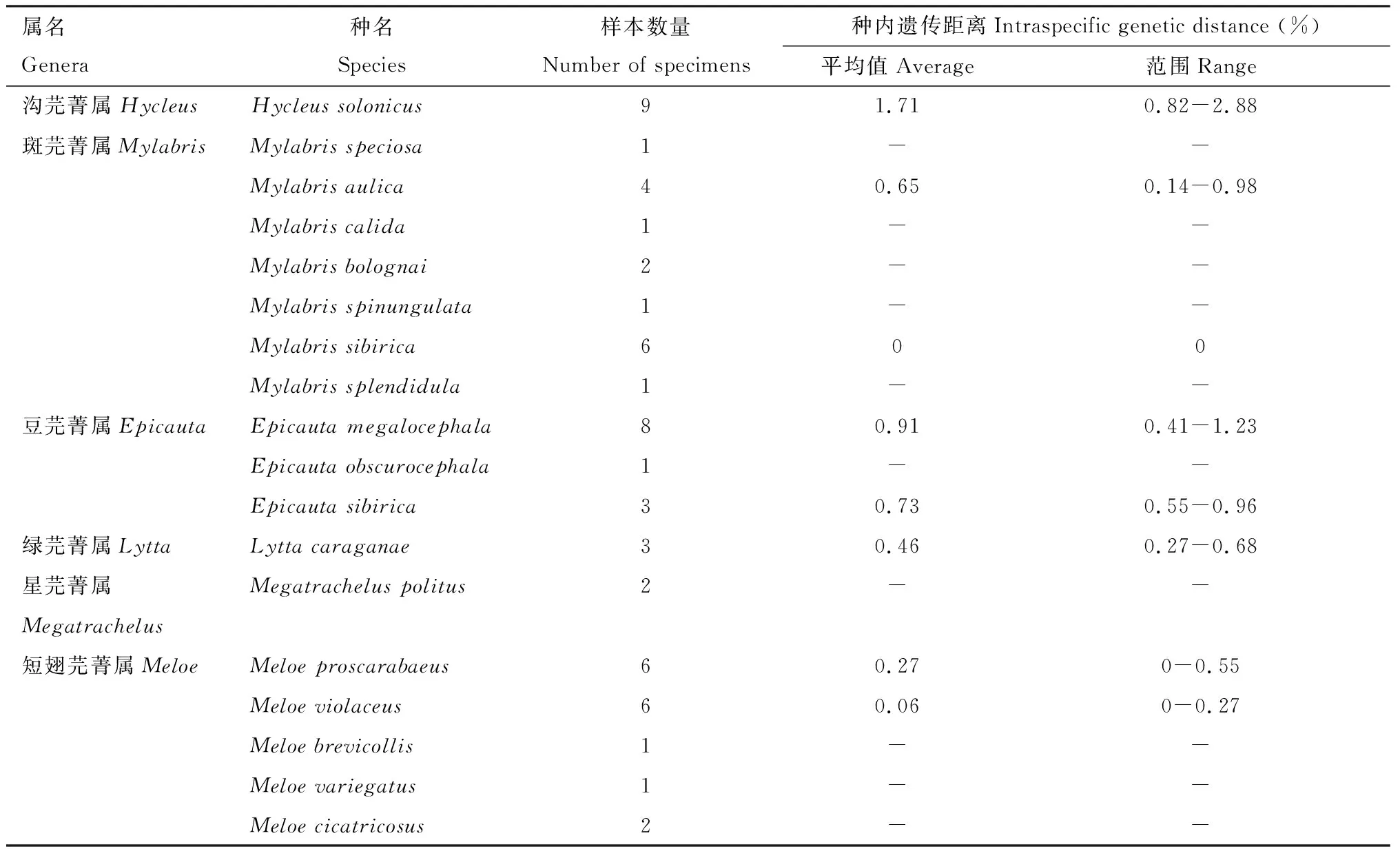
表4 基于CAD基因片段序列计算得出芫菁科种内平均遗传距离和种内遗传距离范围Table 4 Average intraspecific genetic distance and range of intraspecific genetic distance ofMeloidae calculated based on CAD fragment sequences
2.2 系统发育
利用PhyloSuite内嵌的ModelFinder(Kalyaanamoorthyetal., 2017)进行最佳碱基替代模型分析, 实现Akaike’sInformation Criterion (AIC)选择最适模型,3个数据集的最适模型均为GTR+F+I+G4。
对58头芫菁样本进行BI分析,基于COI,CAD和COI+CAD串联序列数据集分别建树,显示出高度相似的拓扑结构(图1),联合数据集分支支持度高于单基因建树,高度支持这18个种的单系性。除Hycleussolonicus,Meloevariegatus,Meloebrevicollis和Epicautaobscurocephala这几个分支的后验概率(posterior probability, PP)<1,其余种划分PP均等于1。
2.3 物种界定
本实验对58个芫菁样本(不含外群)的分子物种界定结果如图1所示,具体如下:
ABGD划分结果包括初始划分(initial partition)和递归划分(recursive partition)两种情况。
基于COI基因片段和COI+CAD串联基因片段序列的划分结果显示,初始划分较为稳定,遗传距离在0.001~0.1之间,被划分为18个分子操作分类单元(molecular operational taxonomic unit, MOTU),支持18个形态种,且与BI树拓扑结构一致。递归划分先验值浮动较大,不够稳定。基于CAD基因片段序列的ABGD划分结果显示,属级分类单元的划分和形态划分吻合,而物种阶元上则将Meloeproscarabaeus和Meloeviolaceus划分为一个MOTU,物种划分紊乱,如图2(A, B, C)。
基于3个数据集的GMYC分析所划分的MOTUs结果皆与形态种相符。单阈值分析,基于COI基因片段序列划分的似然实体数量为19,置信区间为19~22,基于CAD基因片段序列划分的似然实体数量为19,置信区间为1~49,基于COI+CAD串联序列划分的似然实体数量为19,置信区间为15~21,如图3-5所示。
bPTP分析结果表明,基于COI基因序列划分得到21个MOTUs,将Mylabrisaulica,Meloeproscarabaeus和Lyttacaraganae各自分为了2个MOTUs;
基于CAD基因序列划分为17个MOTUs,将Meloeproscarabaeus和Meloeviolaceus归为一个MOTU;
基于COI+CAD基因串联序列得到18个MOTUs,和形态学划分结果一致,如表6-8所示。
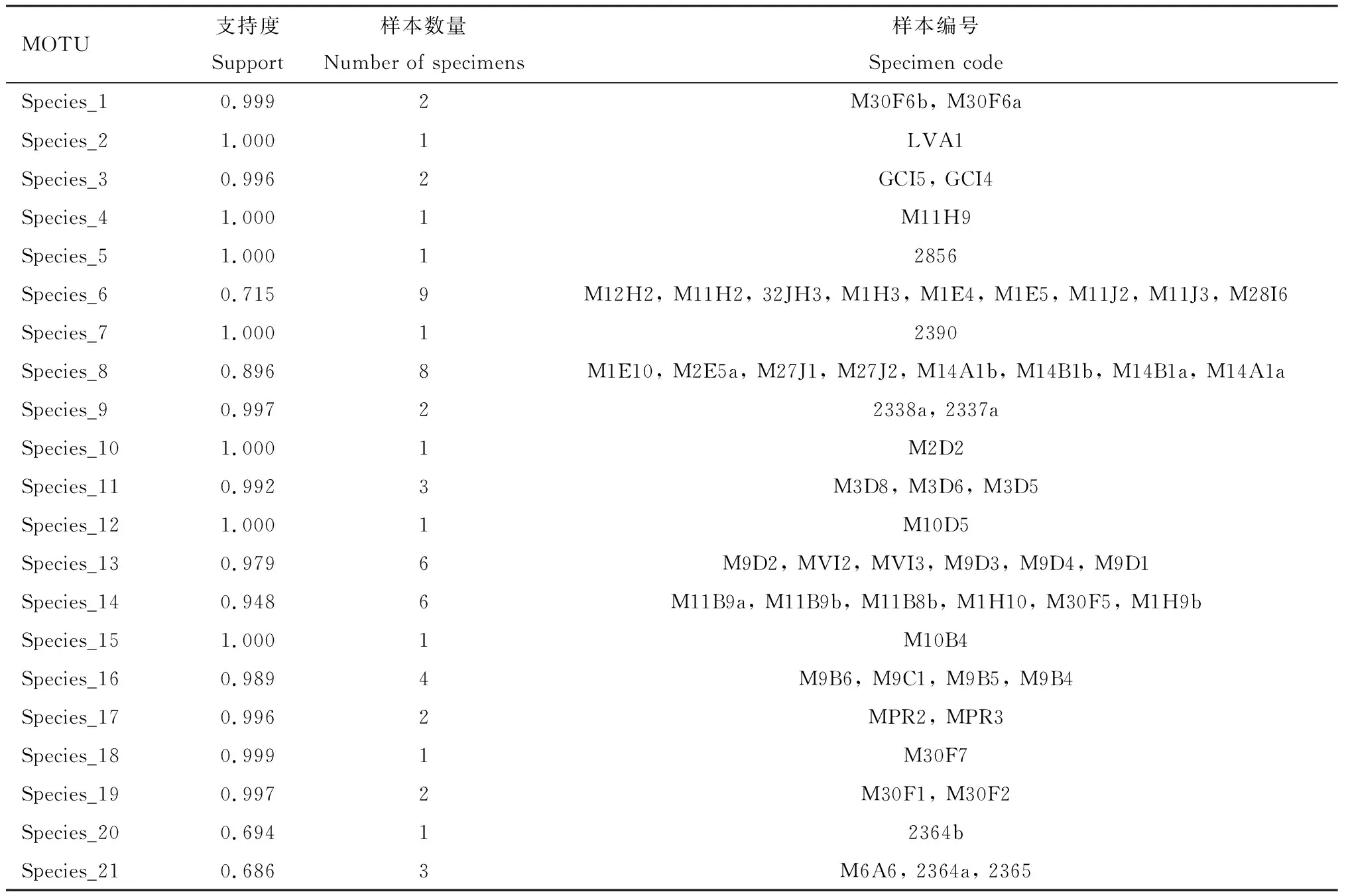
表6 基于COI序列通过bPTP方法得到的芫菁科物种划分Table 6 Species division of Meloidae based on COI sequence by bPTP method
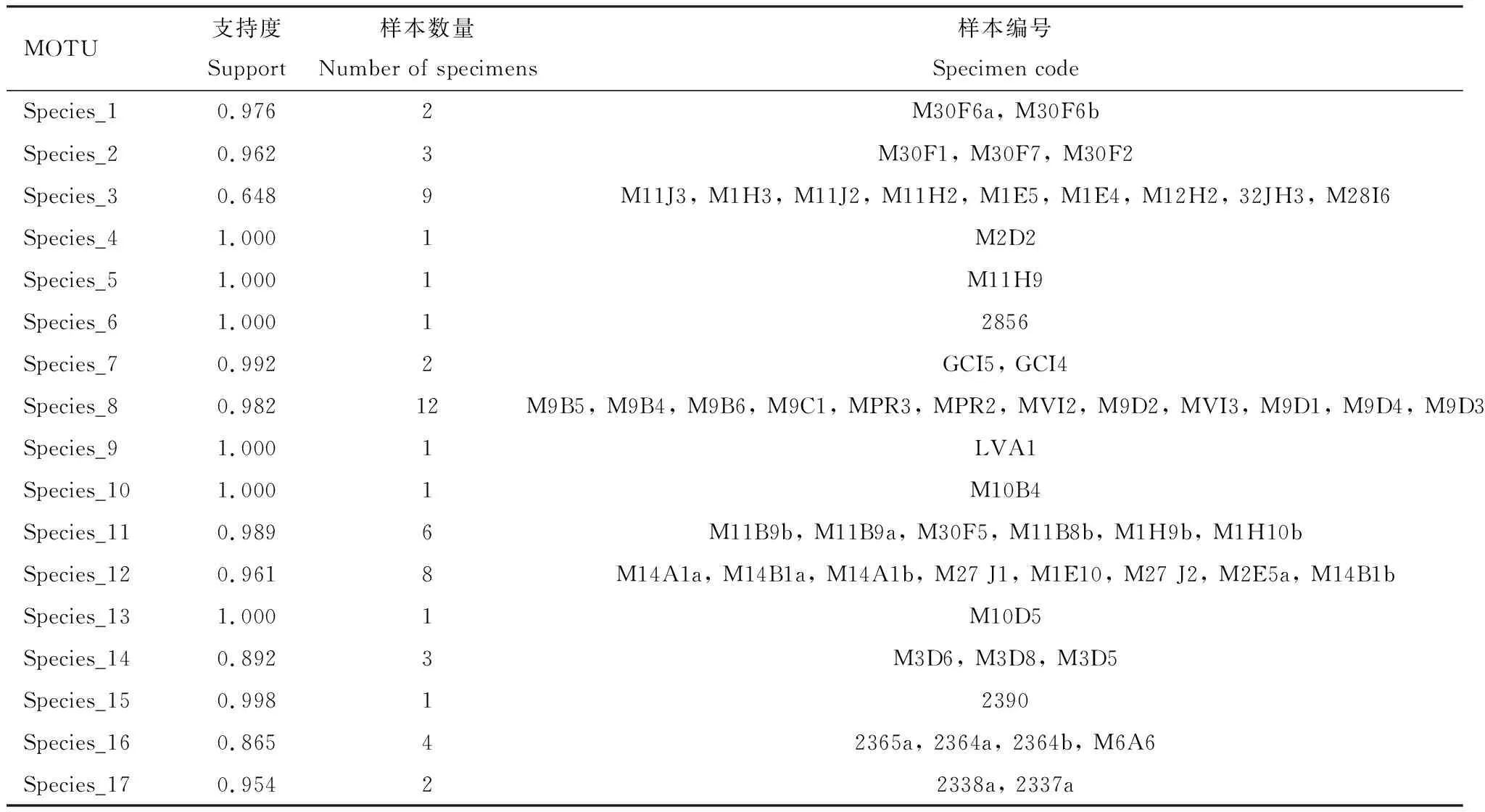
表7 基于CAD序列通过bPTP方法得到的芫菁科物种划分Table 7 Species division of Meloidae based on CAD sequence by bPTP method
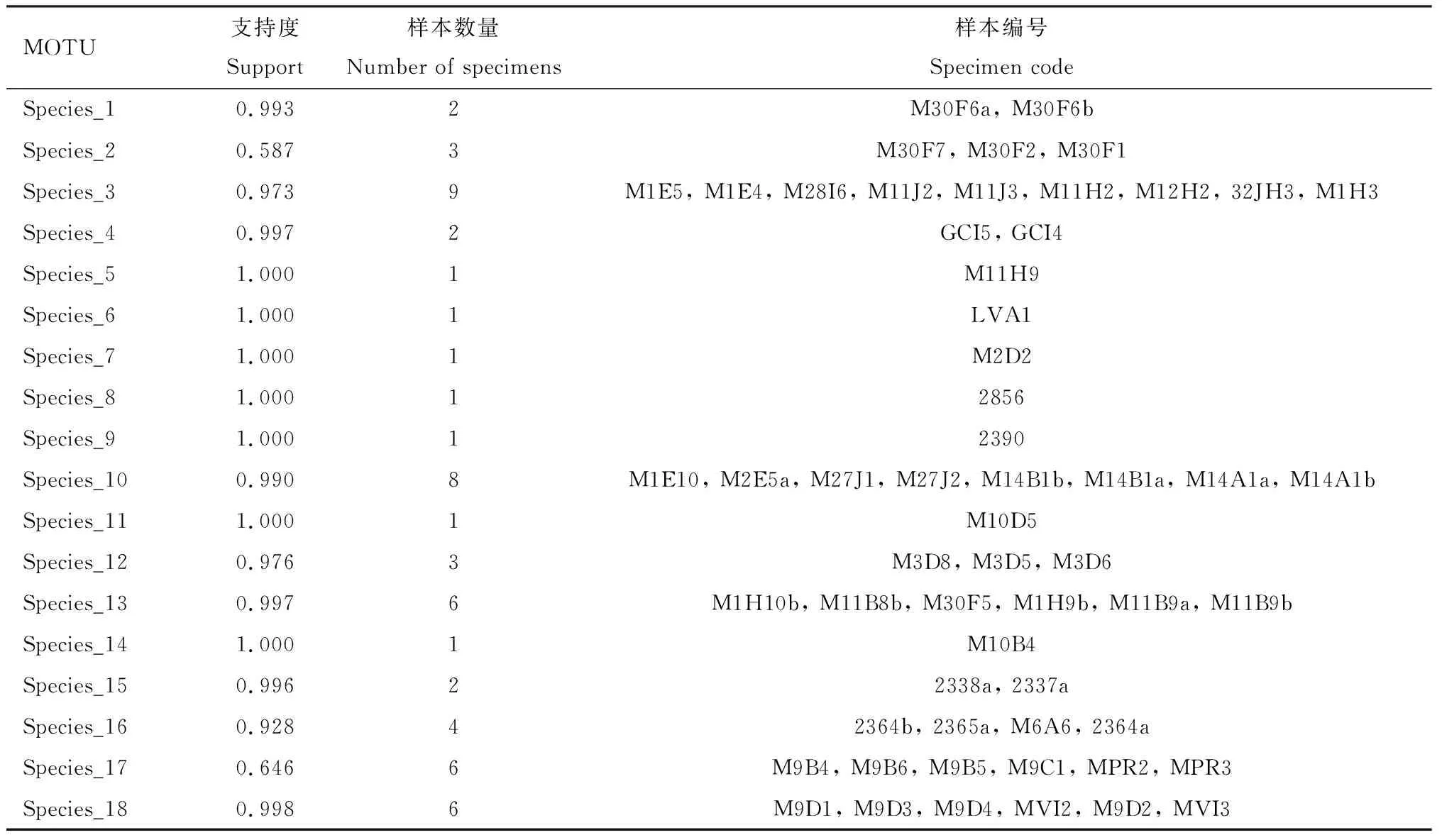
表8 基于COI+CAD串联序列通过bPTP方法得到的芫菁科物种划分Table 8 Species division of Meloidae based on concatenated COI+CAD sequence by bPTP method
BPP是基于给定指导树进行分析,得到18个MOTUs的后验概率(PP)是0.98925,远大于其他划分方式的PP,即支持将58个样本划分为18个独立物种,支持指导树给定的18个物种,与形态学划分一致。
Liu等 (2016)首次应用分子系统学方法对芫菁科昆虫的物种划分问题进行了探讨并提出了2个新异名,然而此工作依据的是外部形态、遗传距离和系统树拓扑结构的综合结果,并未使用其他分子物种界定方法进行分析。
本研究对中国北方常见的芫菁科6属18个形态种进行分子物种界定,总体而言所得结果基本与形态划分一致,仅基于CAD序列的ABGD分析结果和基于单基因的bPTP分析结果有所差异(图1)。该结果一方面验证了形态种划分结果,例如目前使用触角末节形状对具有相似鞘翅斑纹的圆点斑芫菁和苹斑芫菁进行区分(Pan and Ren, 2020),该形态界定结果得到了分子物种界定结果的支持;
另一方面也显现出对芫菁科昆虫相对可靠的可用于分子物种界定的DNA序列和分析方法,即多基因片段串联序列优于COI序列优于CAD序列,ABGD方法最简便、准确性相对较高且稳定,在一定程度上可减少人为界定的误差。在进行ABGD和bPTP分析时,发现基于CAD的划分对物种划分不清晰,因此推测是由于CAD基因进化速率较慢,种间差异小,所以导致种的划分紊乱。
此外,参考分子物种界定结果,基于COI基因种内和种间的遗传距离分析结果表明,在芫菁科中,当遗传距离在1.66%以下可认为是同一物种的种内变异,而大于9.49%则可认为是不同的物种,而在1.66%~9.49%之间则需要综合更多证据谨慎处理(表2和3)。基于CAD基因种内和种间的遗传距离和物种界定综合分析结果表明,在芫菁科中,当遗传距离在1.71%以下可认为是同一物种的种内变异,而大于1.71%则有成为不同物种的可能,在1.71%~2.77%之间则需要综合更多证据谨慎处理(表4和5)。
综上,基于COI和CAD的独立和串联数据集和ABGD, GMYC, PTP和BPP 4种分析方法均适用于芫菁科昆虫的分子物种界定,但有一定的误差,需多基因、多方法综合考虑。此外,形态和分子物种界定方法的结合能够有效提高芫菁科昆虫物种划分的准确性。
猜你喜欢 界定遗传物种 新《著作权法》视域下视听作品的界定社会科学战线(2022年5期)2022-07-23“85后”非遗传承人的旗袍梦华人时刊(2021年21期)2021-03-09我国首次对“碰瓷”作出明确界定汽车维修与保养(2020年11期)2020-11-23还有什么会遗传?动漫界·幼教365(小班)(2019年10期)2019-10-28还有什么会遗传动漫界·幼教365(大班)(2019年10期)2019-10-28还有什么会遗传?动漫界·幼教365(中班)(2019年10期)2019-10-28回首2018,这些新物种值得关注科学大众(中学)(2019年3期)2019-05-17电咖再造新物种汽车观察(2018年10期)2018-11-06世界上的15个最不可思议的新物种科技知识动漫(2017年1期)2017-02-06高血压界定范围金色年代(2016年4期)2016-10-20